根据中国现行的医疗器械监督管理法规,经营第一类医疗器械不需要进行备案。
法规依据
根据国家市场监督管理总局发布的《医疗器械经营监督管理办法》规定,国家对医疗器械按照风险程度实行分类管理。其中,第一类医疗器械风险程度最低,实行常规管理。具体而言,从事第一类医疗器械经营的企业,无需向所在地设区的市级人民政府负责药品监督管理的部门办理经营备案。
经营者的义务与要求
尽管无需备案,但经营者仍需遵守相关法律法规,确保产品安全有效。核心义务包括:
- 合法资质:经营者应当是依法注册的企业或个体工商户,并具备与其经营规模和范围相适应的经营、贮存场所和条件。
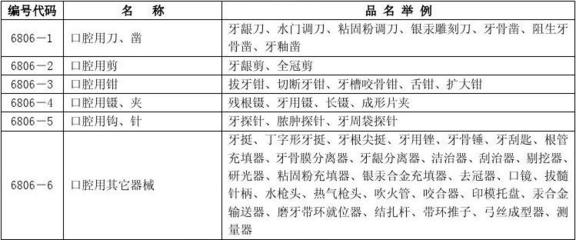
- 产品合法性:所经营的第一类医疗器械必须是已取得医疗器械备案凭证的合法产品。经营者应向供货方索取并查验相关资质和证明文件。
- 质量保障:应建立并执行进货查验记录制度,确保产品来源可追溯、质量有保障。记录应真实、准确、完整,并按规定期限保存。
- 合规经营:不得经营未依法备案、无合格证明文件、过期、失效、淘汰的医疗器械。
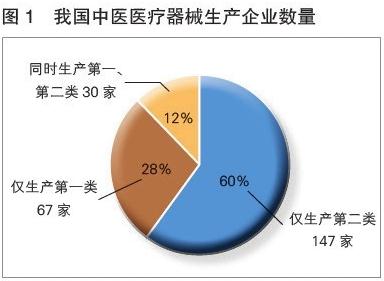
与第二、三类医疗器械的区别
此规定与经营第二类、第三类医疗器械的要求形成鲜明对比:
- 经营第二类医疗器械:需向所在地设区的市级药品监督管理部门办理经营备案。
- 经营第三类医疗器械:需向所在地设区的市级药品监督管理部门申请经营许可,审批要求最为严格。
###
经营第一类医疗器械的门槛相对较低,流程上免去了备案环节,降低了企业的行政成本。但这绝不意味着监管的缺位。经营者必须自觉承担产品质量安全的主体责任,严格遵守《医疗器械监督管理条例》等法规,在便利化的同时守住安全底线。监管部门也会通过日常监督检查、产品抽检等方式进行事中事后监管,确保市场秩序和公众用械安全。
建议:企业在开展经营活动前,务必核实所经营产品的具体分类,可通过查询国家药品监督管理局数据库中的《医疗器械分类目录》进行确认。密切关注相关法规的动态更新,以确保经营活动的持续合规。